A Modular Strategy for Generating Starting Conformations and Data Structures
of Polynucleotide Helices for Potential Energy Calculations
We describe a simple and rapid algorithm for generating data structures
and starting coordinates of polynucleotides for potential energy calculations.
The algorithm is tailored to investigations in cartesian coordinate, rather
than dihedral angle, space. First, instead of a tree structure for molecular
design, we set up a helix from a simple list of bonds for the basic DNA
subunits (sugar, phosphate and bases). Second, instead of using successive
transformations to obtain a set of coordinates in one reference frame,
we apply a simple "matching" routine to patch DNA subunits. Third, we avoid
ring closure and geometry optimization by allowing deviations from
equilibrium values only for P-O3' bond lengths and O5'-P- O3' bond angles
at the residue connection sites. A double-stranded helix is constructed
from duplex building blocks (2 hydrogen-bonded nucleotides) which are in
turn built from the basic structural units. Every building block is constructed
from two sets of geometric variables: {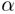 ,
, 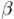 ,
, 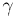 ,
, 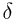 ,
, 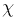 ,
P,
,
P, 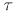 max}, one for
each strand. The building blocks are then assembled into a helix by using
6 rigid body transformations {
max}, one for
each strand. The building blocks are then assembled into a helix by using
6 rigid body transformations {  x, y,z,
x, y,z, 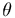 roll, tilt, twist}.
For cartesian space programs, generating starting coordinates by this procedure
is particularly useful as an alternative to using actual crystal structure
coordinates. After describing the algorithm in detail, we illustrate how
it was used to generate model A, B, and Z DNA helices. We conclude
by suggesting how the algorithm can be used to pursue a build-up technique
and to set up a wide range of starting conformations in the goal of locating
novel helical structures.
roll, tilt, twist}.
For cartesian space programs, generating starting coordinates by this procedure
is particularly useful as an alternative to using actual crystal structure
coordinates. After describing the algorithm in detail, we illustrate how
it was used to generate model A, B, and Z DNA helices. We conclude
by suggesting how the algorithm can be used to pursue a build-up technique
and to set up a wide range of starting conformations in the goal of locating
novel helical structures.

 Click to go back to the publication list
Click to go back to the publication list
Webpage design by Igor Zilberman