Long Timestep Dynamics of Peptides by the Dynamics Driver Approach
Previous experience with the Langevin/implicit-Euler scheme for dynamics
("LI") on model systems (butane, water) has shown that LI is numerically
stable for timesteps in the 5-20 fs range but quenches high-frequency modes.
To explore applications to polypeptides, we apply LI to model systems (several
dipeptides, a tetrapeptide, and a 13-residue oligoalanine) and also develop
a new dynamics driver approach ("DA"). The DA scheme, based on LI, addresses
the important issue of proper sampling, which is unlikely to be solved
by small-timestep integration methods or implicit methods with intrinsic
damping at room temperature, such as LI. Equilibrium averages, time-dependent
molecular properties, and sampling trends at room temperature are reported
for both LI and DA dynamics simulations, which are then compared
to those generated by a standard explicit discretization of the Langevin
equation with a 1 fs timestep. We find that LI's quenching effects are
severe on both the fast and slow (due to vibrational coupling) frequency
modes of all-atom polypeptides and lead to more restricted dynamics at
moderate timesteps (40 fs). The DA approach empirically counteracts these
damping effects by adding random atomic perturbations to the coordinates
at each step (before the minimization of a dynamics function). By restricting
the energetic fluctuations and controlling the kinetic energy, we
are able with a 60 fs timestep to generate continuous trajectories that
sample more of the relevant conformational space and also reproduce reasonably
Boltzmann statistics. Although the timescale for transition may be accelerated
by the DA approach, the transitional information obtained for the alanine
dipeptide and the tetrapeptide is consistent with that obtained by several
other theoretical approaches that focus specifically on the determination
of pathways. While the trajectory for oligoalanine by the explicit scheme
over the nanosecond timeframe remains in the vicinity of the full 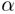 R-helix
starting structure, and a high-temperature (600ºK) MD trajectory departs
slowly from the helical structure,
the DA-generated trajectory for the same CPU time exhibits unfolding and
refolding and reveals a range of conformations with an intermediate
helix content. Significantly, this range of states is more consistent
with spectroscopic experiments on small peptides, as well as the cooperative
two-state model for helix coil transition. The good, near-Boltzmann statistics
reported for the smaller systems above, in combination with the interesting
oligoalanine results, suggest that DA is a promising tool for efficiently
exploring conformational spaces of biomolecules and exploring folding/unfolding
processes of polypeptides.
R-helix
starting structure, and a high-temperature (600ºK) MD trajectory departs
slowly from the helical structure,
the DA-generated trajectory for the same CPU time exhibits unfolding and
refolding and reveals a range of conformations with an intermediate
helix content. Significantly, this range of states is more consistent
with spectroscopic experiments on small peptides, as well as the cooperative
two-state model for helix coil transition. The good, near-Boltzmann statistics
reported for the smaller systems above, in combination with the interesting
oligoalanine results, suggest that DA is a promising tool for efficiently
exploring conformational spaces of biomolecules and exploring folding/unfolding
processes of polypeptides.

 Click to go back to the publication list
Click to go back to the publication list
Webpage design by Igor Zilberman