BIOMOLECULAR DYNAMICS AT LONG TIMESTEPS: Bridging the Timescale Gap Between
Simulation and Experimentation
Innovative algorithms have been developed during the past decade for simulating
Newtonian physics for macromolecules. A major goal is alleviation of the
severe requirement that the integration timestep be small enough to resolve
the fastest components of the motion and thus guarantee numerical stability.
This timestep problem is challenging if strictly faster methods with the
same all-atom resolution at small timesteps are sought. Mathematical techniques
that have worked well in other multiple-timescale contexts-where the fast
motions are rapidly decaying or largely decoupled from others-have nor
been successful for biomolecules, where vibrational coupling is strong.
This review examines general issues that limit the timestep and describes
available methods (constrained, reduced-variable, implicit, symplectic,
multiple-timestep and normal-mode-based schemes). A section compares results
of selected integrators for a model dipeptide, assessing physical and numerical
performance. Included is our dual timestep method LN, which relies on an
approximate linearization of the equations of motion every  t
interval (5 fs or less), the solution for which is obtained by explicit
integration at the inner timestep
t
interval (5 fs or less), the solution for which is obtained by explicit
integration at the inner timestep 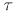 (e.g., 0.5 fs). LN is computationally competitive, providing 4-5 speedup
factors, and results are in good agreement, in comparison to 0.5 fs trajectories.
(e.g., 0.5 fs). LN is computationally competitive, providing 4-5 speedup
factors, and results are in good agreement, in comparison to 0.5 fs trajectories.
These collective algorithmic efforts help fill the gap between the time
range that can be simulated and the timespans of major biological interest
(milliseconds and longer). Still, only a hierarchy of models and methods,
along with experimental improvements will ultimately give theoretical modeling
the status of partner with experiment.

 Click to go back to the publication list
Click to go back to the publication list
Webpage design by Igor Zilberman